氧化应激和新冠关系密切,抗氧化的氢气有没有戏?
严重急性呼吸综合征冠状病毒2 (SARS-CoV-2)引起细胞因子释放综合征(CRS),导致多器官功能障碍。线粒体动力学是抵御环境侵害的基础,但它们对病毒感染非常敏感。线粒体缺陷是活性氧(ROS)的潜在来源。感染SARS-CoV-2会损害线粒体,改变自噬,降低一氧化氮(NO),增加烟酰胺腺嘌呤二核苷酸磷酸氧化酶(NOX)和ROS。2019冠状病毒病(COVID-19)患者表现为激活toll样受体(TLRs)和核苷酸结合和寡聚结构域(NOD-)、富亮氨酸重复序列(LRR-)、含吡咯结构域蛋白3 (NLRP3)炎症小体。SARS‐CoV‐2激活TLRs和NLRP3诱导白细胞介素6 (IL-6)、IL-1β、IL-18和乳酸脱氢酶(LDH)。本文概述了COVID-19的炎症回路及其背后发生的情况,NOX/ROS的相互作用及其在缺氧和血栓形成中的作用,以及ROS清除剂在减少COVID-19相关炎症中的重要作用。
主要参考文献
Anwar M M, Sah R, Shrestha S, et al. Disengaging the COVID-19 Clutch as a Discerning Eye Over the Inflammatory Circuit During SARS-CoV-2 Infection[J]. Inflammation, 2022: 1-20.
本文7600字,供学术探讨。没有耐心读者此处留步。
一、介绍
冠状病毒病-19 (COVID-19)对公共卫生构成威胁,全球有近5亿例病例,约600万人死亡[1,2]。严重急性呼吸综合征冠状病毒2 (SARS-CoV-2)入侵人类肺部,与眼睛、鼻子和嘴巴等不同器官的黏膜相互作用。
患有代谢综合征和糖尿病等共病的老年人往往会出现更严重的COVID-19症状。此外,COVID-19导致的死亡率上升还归因于其他风险因素,如老年、糖尿病、高血压和肾脏疾病。例如,65%以上的COVID-19患者患有糖尿病和心血管疾病,其中63%的患者年龄在60岁以上。
SARS-CoV-2损害线粒体,改变自噬,减少一氧化氮(NO),增加烟酰胺腺嘌呤二核苷酸磷酸氧化酶(NOX)和活性氧(ROS)。在COVID-19中,SARS-CoV-2同时激活toll样受体(toll-like receptor, TLRs)和NOD -、LRR -和pyrin domain-containing protein 3 (NLRP3)炎性小体[4,5,6,7,8,9,10,11,12]。SARS-CoV开放阅读框9b (ORF-9b)操纵人线粒体抗病毒信号分子(MAVS)逃避宿主固有免疫,限制抗炎反应,过量产生ROS[10,13]。NOX蛋白家族在炎症细胞中产生ROS,增强病毒致病性[10,11]。哺乳动物的氮氧化物酶及其亚基包括NOX1-5、p22phox、p67phox和NOXO1,它们在肾脏、心脏和内皮细胞中对血管紧张素II (ATII)反应时升高。这些酶和亚基也参与了COVID-19[14, 15]。感染SARS-CoV-2介导炎症细胞因子和趋化因子,其中atii诱导的白细胞介素-6 (IL-6)的合成通常需要nox衍生的ROS[7]。nox2缺陷的患者和小鼠免疫反应增强,倾向于产生具有低ROS水平的自身抗体[8,9]。SARS‐CoV‐2激活TLRs和NLRP3可诱导IL‐6、IL-1β、IL-18和乳酸脱氢酶(LDH)[4,5,16,17,18,19,20,21,22,23]。
本文综述了COVID-19期间线粒体功能障碍、自噬、NOX、NO、ROS、NLRP3和TLRs的同时有害影响(图1)。此外,我们还提到了ROS清除剂在COVID-19中的潜在作用。
二、COVID-19发病的细胞分子过程
1.一氧化氮介导的炎症ROS途径
NOX信号失调在COVID-19患者的共病包括肥胖、糖尿病、冠状动脉疾病和心力衰竭[24]中很明显。在伴有急性呼吸窘迫综合征(ARDS)的COVID-19患者中,ATII通过ROS、IL-6、肿瘤坏死因子-α (TNF-α)和其他细胞因子增加NOX,导致血管收缩和血栓形成(图2)[25,26]。一氧化氮依赖ROS的生成使TNF-α、转化生长因子-β1 (TGF-β1)、ATII和纤溶酶原激活物抑制剂-1 (PAI-1)升高,这些在COVID-19患者中均升高[24,27,28,29,30]。许多内源性和外源性过程都会产生ROS,如NOX、电子传递链、黄嘌呤氧化酶、吸烟、重金属、药物、加工肉类和辐射(图2)[31]。血管平滑肌中干扰素γ (IFN-γ)和ATII触发NOX1表达,缺氧/缺血和TNF-α刺激NOX4的表达[32,33]。内小体NOX2产生促炎白三烯B4 (LTB4),增加病毒介导的致病性中IL-6和ROS的水平[10,34,35,36,37,38]。例如,在没有NOX2的情况下,流感A病毒引起的肺损伤明显减少,这表明NOX2介导的ROS能够刺激病毒感染[35,39]。在COVID-19中,SARS-CoV-2上调ACE和ATII,从而激活吞噬细胞,代谢血红蛋白,导致高铁血症产生羟基自由基(•OH),增加炎症和血栓形成的可能性(图3)[40,41,42,43,44,45,47,47,48,49,50,51]。
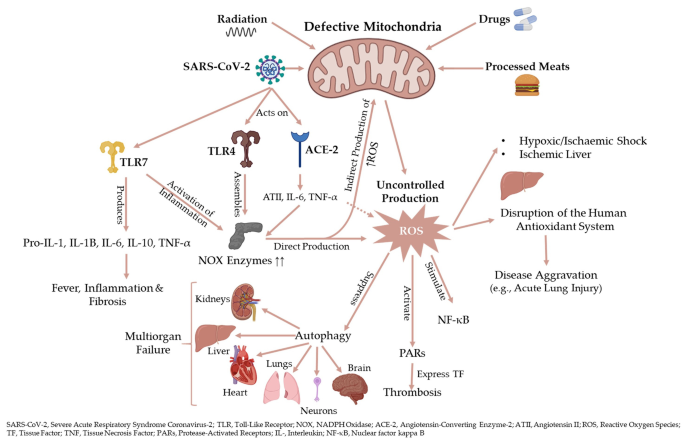
图2 炎症和氧化应激相互作用
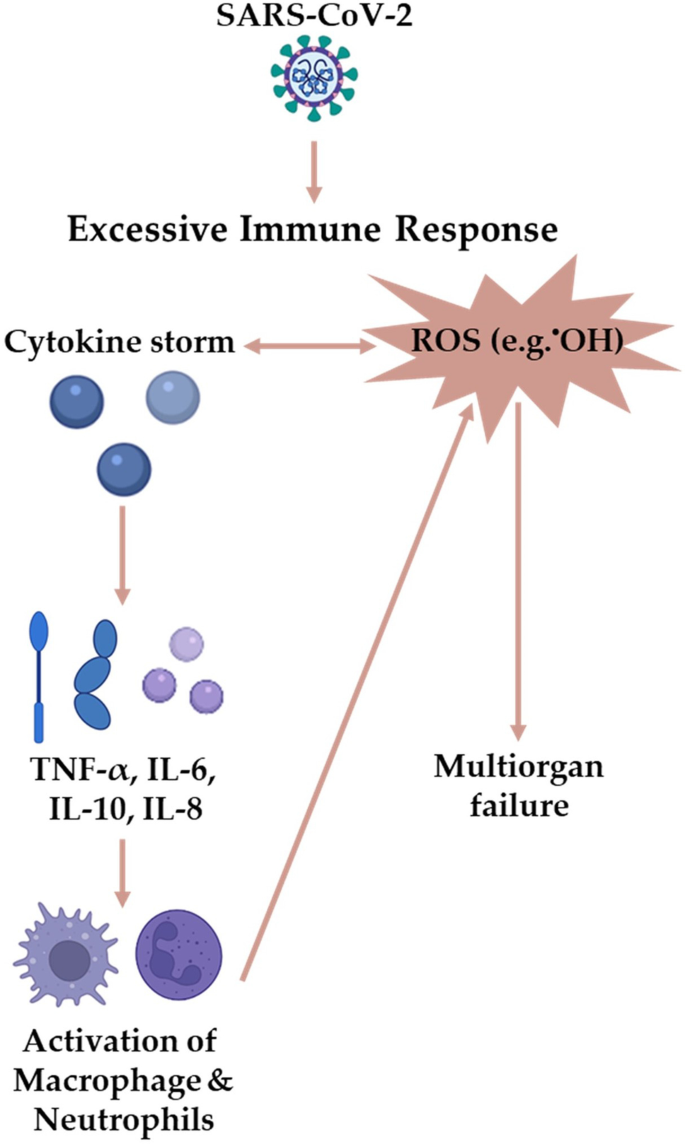
图3 新冠状肺炎病理过程炎症和氧化应激的关系
•OH的形成与氧化应激产物如DNA的4-羟基壬烯醛和丙二醛鸟嘌呤加合物相关,这些产物也是与COVID-19血脂异常相关的磷脂自由基氧化的产物[52,53,54,55]。活性氧与脂类、碳水化合物、蛋白质和核酸相互作用,导致其功能永久性破坏或改变[56]。羟基自由基是最具活性和毒性的活性氧,通过与DNA、碳水化合物、蛋白质和脂类的强烈相互作用导致严重的细胞损伤[57,58,59,60]。不同疾病(如衰老和帕金森病)中的血色沉着症已引起关注,因为铁催化•OH的形成[61,62,63,64]。羟基自由基直接与DNA的所有成分,如嘌呤和嘧啶碱基、脱氧核糖糖骨架等发生反应,引起DNA链断裂中的单链断裂和双链断裂以及碱基或核苷酸的化学修饰[60,65,66]。ROS的不受控制的产生对感染性、炎症性和许多慢性疾病有重要作用。这一证据支持了目前的假设,即NOX是COVID-19发病机制中的一个重要调节因子,阻断NOX的表达可能会阻碍atii诱导的ROS和IL-6的产生,从而减少炎症和组织损伤(图2)。
2.NLRP3的炎症作用
COVID-19患者死后组织显示活性NLRP3炎症小体及其产物,包括IL-1β、IL-18和LDH[16,17,18,19,20,21,22,23]。急性和慢性呼吸系统疾病、创伤性脑损伤、急性肾损伤(AKI)和慢性肾脏疾病(CKD)也报道了NLRP3炎性小体的参与[67]。病毒感染、代谢异常、组织损伤和线粒体功能障碍会产生ROS(如•OH),激活NLRP3炎症小体,触发促炎细胞因子的产生[68,69,70,71,72,73]。幸运的是,线粒体靶向抗氧化剂如分子氢(H2)可以抑制线粒体•OH的产生,从而抑制NLRP3炎症小体、caspase-1和IL-1β的表达[74]。分子氢是一种有效的清除剂,可选择性清除•OH,且对人体无不良影响[75]。最近的一项多中心试验显示,吸入氢氧混合气体可减少covid -19相关的急性和慢性炎症[76]。在急性胰腺炎小鼠模型中,腹腔内富含h2的生理盐水可抑制NLRP3炎症小体的活化、活化B细胞kappa-轻链增强子(NF-κB)的活性以及TNF-α和IL-1β的产生。此外,富h2盐水可提高大鼠存活率,改善肠道损伤和炎症反应,水肿和细胞凋亡可改善肠缺血/再灌注介导的凝血障碍。富氢生理盐水可抑制外周血单个核细胞(PBMCs)中NF-κB和NLRP3炎症小体的活化[77]。因此,H2可能通过抑制NLRP3级联和促炎细胞因子的释放来减少sars - cov -2诱导的炎症反应。
3.NO /ROS失衡
COVID-19引起的持续炎症会扰乱一氧化氮(NO)/ROS平衡,导致多器官衰竭[78]。COVID-19患者及常见共病(如高血压和糖尿病)患者内皮细胞NO明显降低,提示其与急性肺损伤(ALI)和NO/ROS失衡密切相关[79,80,81,82,83,84,85]。严重急性呼吸综合征冠状病毒2可下调血管紧张素转换酶2 (angiotensin-converting enzyme 2, ACE2)的表达,产生促炎细胞因子和ROS,引起过度炎症反应,并通过引起内皮细胞凋亡降低NO水平(图4)[86,87,88,89]。由于ACE-2受体的高表达,SARS-CoV-2颗粒很容易结合其蛋白尖刺进入细胞。因此,代谢健康受损的人更容易出现COVID-19和共病[3]。重症COVID-19病人表现出过度的线粒体活性氧导致线粒体功能障碍,减少生产和生物利用度的核转录因子的激活kappa-light-chain-enhancer激活B细胞(NF-kB), AP-1以及超表达的细胞因子和粘附分子(图2)(90、91、92)。NO供体S-亚硝基-n -乙酰青霉胺(SNAP)显著抑制SARS-CoV-1 ORF1a编码的半胱氨酸蛋白酶和子代病毒S蛋白的膜融合,使病毒在VeroE6细胞中的复制降低> 80%[93,94,95,96,97]。SARS-CoV-2和SARS-CoV在刺突蛋白的受体结合域表现出高度的相似性[98,99]。因此,吸入NO可预防SARS-CoV-2感染,或治疗轻、中、重度COVID-19患者,并可作为机械通气患者的辅助治疗(图4)[83,100,101]。
4.线粒体功能障碍与自噬
在COVID-19期间,缺氧和其他炎症介质损害线粒体的功能[102,103]。线粒体功能障碍是ROS的潜在来源,可影响健康线粒体并促进细胞死亡[104]。线粒体已经成为维持细胞内稳态、代谢、先天免疫反应和决定病毒感染严重程度的关键动态细胞器[105]。线粒体动力学,如融合、裂变和线粒体噬保护环境的损害;尽管如此,由于病毒蛋白或生理改变(如Ca2+稳态破坏、内质网应激、氧化应激和缺氧),它们易受病毒感染[106,107,108]。通过干扰线粒体,病毒扭曲线粒体功能,为病毒增殖创造有利的应激环境(即,线粒体ATP和ROS分别低量和高量),并阻碍线粒体相关的抗病毒信号传导[109]。有缺陷的线粒体是ROS的潜在来源,也可能导致健康的线粒体受损。因此,干扰功能障碍线粒体的快速清除会产生更高水平的ROS,促进细胞死亡[102,104,110,111]。之后,病毒(如SARS-CoV-2)开始通过改变NLRP3炎症小体和自噬等潜在靶点增殖和繁殖[112]。
在covid -19相关的败血症中,sars - cov -2-宿主相互作用释放细胞因子风暴,最终导致多器官衰竭[113]。促炎细胞因子TNF-α增加了人脐静脉内皮细胞(HUVECs)线粒体损伤介导的线粒体ROS[114]。同样,COVID-19显著上调TNF-α以及其他细胞因子和趋化因子(图1和图3)。因此,SARS-CoV-2可能通过上调TNF-α并导致线粒体超微结构异常产生更多ROS来抵消抗病毒反应[115]。病毒通过改变自噬、线粒体自噬和细胞代谢来调节线粒体介导的抗病毒免疫反应,以促进其增殖[112]。
自噬是sars - cov -2介导的COVID-19的一个重要靶点[112]。SARS-CoV对细胞自噬的抑制可能进一步阐明了线粒体功能障碍在COVID-19发病过程中的病理生理作用。细胞通过自噬(即自毁机制),通过隔离膜的启动和延伸、自噬体的形成、自噬体-溶酶体的融合和降解,去除功能失调和多余的细胞成分[112]。线粒体通过产生ROS来调节自噬去除有害成分,而自噬通过线粒体自噬来控制线粒体内稳态[116,117]。病毒感染导致的正常自噬缺失导致线粒体功能障碍和ROS生成(图2)[118]。心血管、神经退行性疾病、慢性肝肾疾病也证实了自噬恶化、线粒体功能障碍和ROS生成之间的相互作用[119,120,121,122]。这些数据支持这样一个事实,即正常自噬的丧失可能是SARS-CoV-2感染扰乱线粒体内稳态的主要原因之一。然而,大量研究报道SARS-CoV、SARS-CoV-2、中东呼吸综合征冠状病毒(MERS-CoV)和小鼠肝炎病毒(MHV)诱导和抑制自噬。进一步调控自噬(即诱导或抑制自噬)的研究将阐述对SARS-CoV-2治疗的影响[123 124、125、126、127、128、129、130、131、132]。
5.自噬缺失和ROS
新冠肺炎老年患者表现出脆弱的抗氧化防御能力和夸张的氧化损伤。COVID-19患者的ARDS发病需要激活“ROS机制”并结合先天免疫促进NF-κB,加剧促炎宿主反应(图2)[133]。在体外和体内的SARS-CoV发病、严重程度和呼吸道疾病进展过程中,ROS的过量产生显著干扰了抗氧化系统[134,135]。人类也会经历与年龄相关的自噬损失或暴露在ROS中。自噬可能与衰老表型有关,表明衰老改变了适应性免疫反应和宿主的促炎状态[136]。例如,老年小鼠比年轻小鼠严重地经历了sars - cov诱导的肺损伤[137]。由于NF-kB作为核心角色的基因表达差异,老年猕猴上调病毒-宿主炎症反应[137]。老年患者多叶病变的发生率也明显高于中青年患者[138]。在老年患者中,自噬抑制导致线粒体功能障碍的同时下降和易感的合并症,可能解释了为什么与年轻患者相比,老年COVID-19患者会表现出严重的临床表现,最终导致多器官衰竭(图2)。世界卫生组织宣布,目前批准的药物(如氯氮平、格列本脲、卡培戊烷)可用于治疗COVID-19,通过靶向NLRP3炎症小体和自噬来抑制SARS-CoV-2的增殖[139,140,141,142,143]。
6.TLRs、NOX和ROS之间相互影响
有证据支持NOX、ROS、炎症介质与SARS-CoV-2发病机制之间的关联,以及在TLR4/NOX相互作用期间,ROS信号与TLR4激活之间的关系(图2)[144,145]。二苯醚氯化碘(diphenyleneiodonium chloride, DPI)可抑制酒精性脂肪肝损伤中TLR2、4和9的上调[146]。人类细胞强调了NOX2抑制剂在病毒感染中的潜在作用。在呼吸道合胞病毒、鼻病毒和人类免疫缺陷病毒(HIV)中,TLR7激活NOX2产生ROS,并修饰TLR7的单个半胱氨酸残基,抑制关键的抗病毒和体液信号转导[147]。合胞病毒细胞质组分识别TLR7和其他传感器分子;线粒体产生大量氧化线粒体DNA的·OH,驱动从NLRP3级联到释放促炎细胞因子(图2)[72,148]。
严重急性呼吸综合征冠状病毒2结合TLRs激活并调节pro-IL-1、NLRP3、IL-1β、IL-6、IL-10和TNF-α。这种级联会导致肺部炎症和纤维化,这表明TLR通路在SARS-CoV感染中是一种保护机制[149,150,151]。toll样受体(如TLR3、4、7、8和9)识别了许多病毒保守模式,其中髓样分化初级反应88 (MyD88) - TLR通路的重要组成部分-在中性粒细胞和巨噬细胞中组装NOX产生ROS(图2)[152,153]。髓样分化初级反应88激活包含tir结构域适配器诱导干扰素(TRIF)依赖的信号通路,从而激活IFN-1、NF - kB和丝裂原活化蛋白激酶(MAPK)通路[154]。TLR-MyD88下游信号通路和NF-kB的激活是SARS-CoV感染的一个标志,抑制NF-kB可显著降低呼吸道冠状病毒感染并提高小鼠的存活率[151,155]。
sars - cov恢复期患者线粒体和ros响应基因上调[144]。例如,ROS/NF‐kB/TLR(主要是TL4)信号通路在SARS-CoV触发时导致ALI。TLR4-TRIF-TRAF6致病途径介导ALI的严重程度。TLR4或TRIF表达缺失保护小鼠免受h5n1诱导的ALI,提示ALI的严重程度取决于ROS和先天免疫。
三、NOX / ROS在介导COVID-19缺氧、缺血损伤、血栓形成和纤维化中的潜在作用。
COVID-19细胞因子风暴期间发生的严重缺氧是导致心肌和肝脏损伤、中毒性脑病、四肢缺血和凝血异常的主要原因[156,157,158,159]。虽然在肺内皮中NOX的活化可介导缺血介导ROS的增加,但仍缺乏数据支持NOX家族在COVID-19患者缺氧/缺血中的作用[160,161]。冠状动脉结扎小鼠模型显示,NOX2可导致心脏的不良损伤[162]。鼻病毒、SARS-CoV和人血小板缺氧可在体外产生nox2依赖性ROS[163]。NOX2基因缺失可抑制小鼠间歇性缺氧和氧化应激引起的认知功能缺损[164,165]。移植p47phox缺陷骨髓的小鼠肺缺血和促炎细胞因子水平降低[166]。罗布林- nox2抑制剂降低绵羊血管通透性,在不同的小鼠模型中流产缺血肺和肝损伤、细胞坏死和组织损伤、细胞因子释放和ROS产生[167,168,169,170,171,172,173]。这些数据强调,抑制NOX、ROS或p47phox有望设计有效分子来限制COVID-19患者的缺血损伤[7,103,174]。
1.脑缺血
COVID-19患者出现缺血性中风。缺血性脑卒中占所有卒中的80%以上,其发生是由于大脑中动脉阻塞导致血流立即停止[175,176]。ROS的过量产生会加重氧化应激,导致缺血时的脑损伤,提示降低ROS可能有助于脑卒中的治疗(图1)[177,178,179,180]。研究表明,NOX1、NOX2、NOX4和NOX5与脑功能障碍和ROS释放相关[181,182,183,184,185,186]。NOX2基因缺失对大脑中动脉闭塞(MCAO)模型脑卒中有保护作用。功能性NOX2缺失和NOX2敲除(KO)小鼠在脑缺血时水肿、病变体积、血脑屏障(BBB)渗漏、缺血后炎症基因表达和氧化应激标志物显著减少,神经功能改善[187,188,189,190]。nox2缺乏的海马神经元视网膜缺血模型小鼠暴露于氧/糖剥夺(OGD)时,ROS水平较低,神经元细胞死亡减弱[191]。因此,脑卒中的治疗应采取有效的NOX抑制策略,尤其是NOX2。然而,广泛的模拟人类生物系统的研究对于验证来自体内模型的数据至关重要,这些模型给出了小器官和损伤组织中相对较大的半暗带。
2.血栓形成和纤维化
据报道,COVID-19患者出现微血栓、肺栓塞、内皮衰竭和弥散性血管内凝血(DIC)[7,192,193,194,195,196]。病毒通过线粒体ROS介导的蛋白酶激活受体(PAR1和PAR2)激活凝血通路,过度产生促炎细胞因子[196,197,198,199,200,201]。血小板、内皮细胞和上皮细胞以及血管和非血管平滑肌上表达的PAR1/ par2都参与炎症反应[202,203,204,205]。内皮细胞中NOX亚基p22phox的上调产生ROS,促进PAR1和par2介导的组织因子(TF)诱导,引起急性和慢性炎症(图2)[206,207,208,209]。在炎症过程中,铁(III)产生•OH,将可溶性血浆纤维蛋白原转化为异常的纤维蛋白凝块,形成致密的、不受酶降解(即抗凝血)的沉积(图4)[210211212]。炎症过程中,组织纤溶酶原激活物(tPA)可下调内皮细胞中的IL-1α和IL-1β[213,214]。3例机械通气的COVID-19患者表明,tPA对ARDS有治疗作用,显示动脉氧分压/分次吸氧比有短暂改善[215]。然而,这种改善在治疗结束后失去,因为依赖于nox的ROS会抑制tPA活性,导致血栓形成[216,217]。
COVID-19死亡患者肝、肺损伤活检显示严重的炎症反应,IL-2、IL-6、IL-8水平较高。IL-10和IFN-γ[218]。PAR1和PAR2的激活可触发纤维化反应,诱导NF-kB和IL-6、IL-8和MCP-1的释放,这些有助于SARS-CoV-2感染期间白细胞募集(图1)[219]。在慢性肝病和肺纤维化中,PAR(即PAR2)的直接上调可增加ROS的产生,通过诱导肝细胞凋亡、气道阻塞和肺水肿来促进纤维生成[209,220,221,222,223]。这与par -2缺陷小鼠表现出炎症减轻和生存率提高的事实是一致的[224,225]。因此,我们预计,依赖par -2的ROS可能会导致COVID-19患者的肺和肝损伤,特别是在肝病等易感疾病中,导致免疫抑制和疾病侵袭[25,226,227]。
四、活性氧清除剂能有效对抗covid-19吗?
天然化合物如番茄红素、多酚、槲皮素、根皮素、小檗碱和萝卜硫素显示出预防SARS-CoV-2感染的潜力[228,229,230,231]。卵磷脂化超氧化物歧化酶(PC-SOD)具有良好的生物利用度、安全性(已在I期和II期研究中证实),并具有降低COVID-19氧化应激危害的调节作用[232,233,234,235,236]。与非卵磷脂形式的酶相比,它是一种长寿命和高生物利用度的合成产品[237,238]。例如,静脉注射PC-SOD是安全的,可以抑制肺气肿和纤维化、肺炎症或ARDS、蛋白酶的激活以及体外和动物模型的表达[234,239,240,241,242,243]。卵磷脂化超氧化物歧化酶可降低III-IV期特发性肺纤维化患者血清LDH和表面活性蛋白A,且无明显副作用。如果在疾病过程中尽早使用,它将发挥更大的肺部保护作用[233]。
五、结论与未来发展方向
本文综述了线粒体功能障碍、NOX、ROS、NLRP3炎症小体、TLRs和NO作为COVID-19“炎症回路”的密切关系。缺乏正常的自噬会导致线粒体功能障碍和ROS产生等核心问题。随后,自噬和SARS-CoV-2之间可能存在相互作用,但这种相互作用的确切性质仍不清楚。
在SARS-CoV-2感染过程中ROS和NOX之间的串扰,明确地构成了一个新兴的COVID-19分子分析和药物设计路线。其他可能的干扰信号通路(如PAR、TLR- myd88、ROS/NF - kB/TLR和TLR4/TRIF/TRAF6)在SARS-CoV-2发病过程中发生。冠状病毒蛋白酶,特别是3c样蛋白酶(Mpro或3CLpro)是有吸引力的抗病毒药物靶点,因为它们对冠状病毒复制至关重要。这类抗病毒药物将抑制病毒复制和受感染细胞中可能导致健康细胞[6]死亡的信号级联失调。
未来的研究可能揭示COVID-19、SARS-CoV-2 -宿主相互作用期间的线粒体天然抗病毒信号,以及SARS-CoV-2如何利用线粒体形态生理学的改变来造福自身[244,245]。NOS和ROX的抑制剂可能是有希望减少sars - cov -2相关的细胞因子反应、血管高通透性、微血栓形成、组织损伤/缺血和纤维化以及多器官衰竭过程中的高炎症状态的化合物。然而,氮氧化物和活性氧在正常生理中的基本功能应该被考虑。抗氧化剂的使用可能会面临潜在的挑战,如生理干扰、NOX/ROS的生物功能、缺乏目标通路以及无法达到足够的ROS浓度。
能够看出,本文主要是对氧化应激和炎症关系进行了比较好的阐述。对抗氧化物用于这种疾病治疗只是提出了设想,研究证据目前还不够充分。我们的读者都知道,氢气用于这种疾病,已经有比较多个案报道和病例分析研究,但缺乏RCT研究证据。因此目前我们不能声称氢气治疗这种疾病有效。不能声称不是否定其作用,是因为证据不够硬。
在这种情况下,我们唯一能干的事,就是耐心等待证据。证据说没有,我们就说没有,证据说有多少,我们就说有多少,证据说可以我们就说可以,说不可就不可。因为,医学靠证据,不靠理论。再天花乱坠的理论都不能在没有证据的情况下声称。



